
One of many shared, elementary targets of most chemistry researchers is the necessity to predict a molecule’s properties, similar to its boiling or melting level. As soon as researchers can pinpoint that prediction, they’re capable of transfer ahead with their work yielding discoveries that result in medicines, supplies, and extra. Traditionally, nonetheless, the standard strategies of unveiling these predictions are related to a big value — expending time and put on and tear on tools, along with funds.
Enter a department of synthetic intelligence generally known as machine studying (ML). ML has lessened the burden of molecule property prediction to a level, however the superior instruments that almost all successfully expedite the method — by studying from present knowledge to make fast predictions for brand new molecules — require the consumer to have a big degree of programming experience. This creates an accessibility barrier for a lot of chemists, who could not have the numerous computational proficiency required to navigate the prediction pipeline.
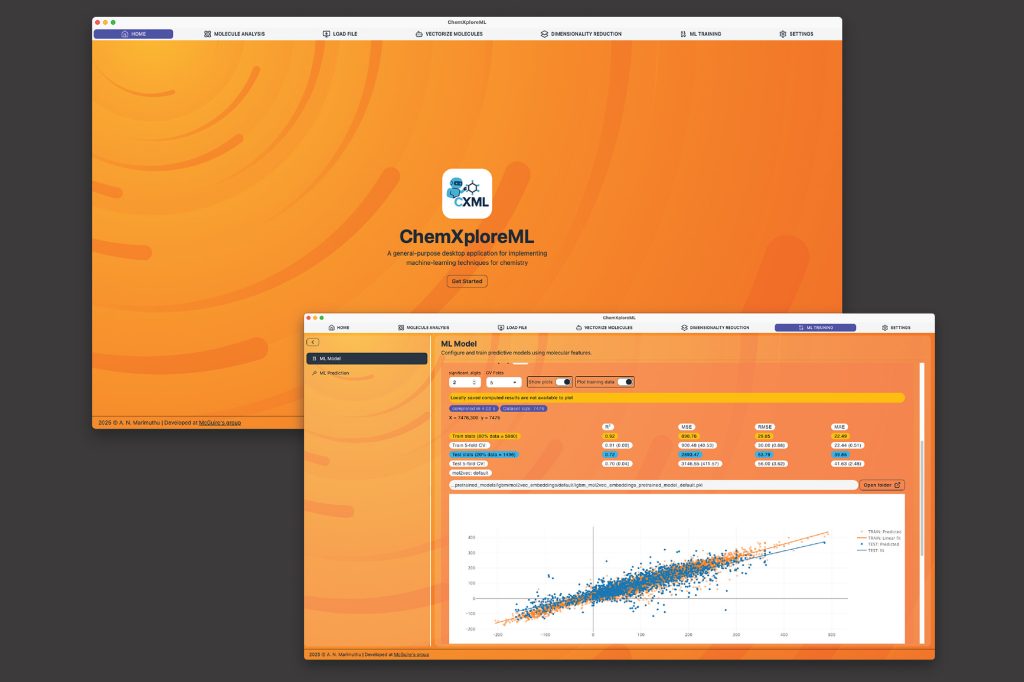
To alleviate this problem, researchers within the McGuire Analysis Group at MIT have created ChemXploreML, a user-friendly desktop app that helps chemists make these crucial predictions with out requiring superior programming expertise. Freely out there, straightforward to obtain, and purposeful on mainstream platforms, this app can be constructed to function completely offline, which helps maintain analysis knowledge proprietary. The thrilling new know-how is printed in an article printed not too long ago in the Journal of Chemical Info and Modeling.
One particular hurdle in chemical machine studying is translating molecular buildings right into a numerical language that computer systems can perceive. ChemXploreML automates this complicated course of with highly effective, built-in “molecular embedders” that rework chemical buildings into informative numerical vectors. Subsequent, the software program implements state-of-the-art algorithms to determine patterns and precisely predict molecular properties like boiling and melting factors, all via an intuitive, interactive graphical interface.
“The purpose of ChemXploreML is to democratize the usage of machine studying within the chemical sciences,” says Aravindh Nivas Marimuthu, a postdoc within the McGuire Group and lead writer of the article. “By creating an intuitive, highly effective, and offline-capable desktop software, we’re placing state-of-the-art predictive modeling instantly into the palms of chemists, no matter their programming background. This work not solely accelerates the seek for new medication and supplies by making the screening course of sooner and cheaper, however its versatile design additionally opens doorways for future improvements.”
ChemXploreML is designed to to evolve over time, in order future strategies and algorithms are developed, they are often seamlessly built-in into the app, guaranteeing that researchers are all the time capable of entry and implement essentially the most up-to-date strategies. The appliance was examined on 5 key molecular properties of natural compounds — melting level, boiling level, vapor stress, crucial temperature, and demanding stress — and achieved excessive accuracy scores of as much as 93 % for the crucial temperature. The researchers additionally demonstrated {that a} new, extra compact methodology of representing molecules (VICGAE) was practically as correct as commonplace strategies, similar to Mol2Vec, however was as much as 10 occasions sooner.
“We envision a future the place any researcher can simply customise and apply machine studying to resolve distinctive challenges, from growing sustainable supplies to exploring the complicated chemistry of interstellar area,” says Marimuthu. Becoming a member of him on the paper is senior writer and Class of 1943 Profession Growth Assistant Professor of Chemistry Brett McGuire.